


 Overview
Overview
Individual lysosomal storage disorders are classified as rare genetic disorders. However, when taken as a group, they are far more common.
The prevalence rate is estimated to be 1 per 7,700 births in some countries (Meikle, PJ et al, Prevalence of Lysosomal Storage Disorders, JAMA 1999; 281:249-254)
Frequently Asked Questions
Which NTSAD diseases are lysosomal storage disorders?
Tay Sachs, Sandhoff, GM1, Fabry, Gaucher, Niemann-Pick, Pompe are lysosomal storage disorders. Tay Sachs and Sandhoff are both categorized as GM-2 gangliosidoses. This is because they are both caused by the build up of GM-2 gangliosides, as described below.
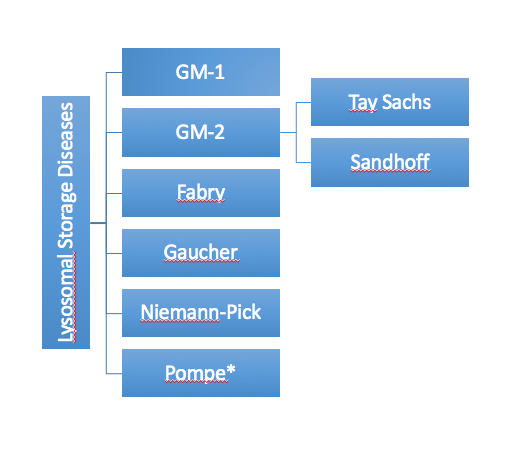
*Pompe is also a glycogen storage disorder.
What is a lysosome and what is a lysosomal storage disorder?
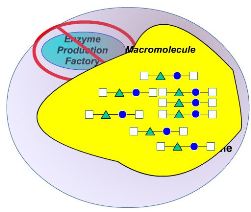
Lysosomes are the cell’s recycling center. They contain digestive enzymes; responsible for breaking down debris in the cell into reusable parts.
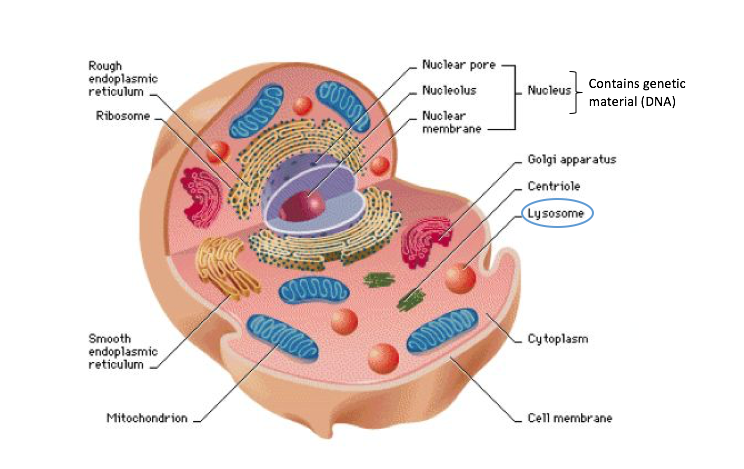
The Cell. Image from: https://awbionotes.wordpress.com/tag/animal-cell/
A lysosomal storage disorder occurs when one of these enzymes is not functioning correctly. When the enzyme does not function correctly, there is a build up of undigested debris (substrate) in the lysosome. This substrate is usually a fatty substance called a sphingolipid. This build up eventually leads to cell degeneration. This then results in the accumulation of substrate in various tissues and organs of the body, causing these organs to function less efficiently, resulting in progressive deterioration in physical and/or mental state, and eventually death.
A visual representation:
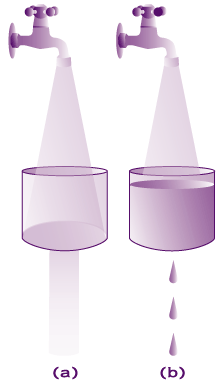
a) In most individuals the substrate (water) can be degraded efficiently by adequate enzyme volume (hole in sink).
b) In affected individuals the amount of enzyme is insufficient to efficiently degrade the substrate and it accumulates. It is this accumulation that causes the disease.
Why does the enzyme not function correctly?
The enzyme is made incorrectly if both copies of the gene that codes for the enzyme contain a spelling error, which is called a mutation. Mutations in the gene can change the way the enzyme is made, and it therefore is unable to do its job. Sometimes the mutations in the gene cause the enzyme to not be made at all. When the enzyme cannot do its job, the substrate builds up.
Which enzyme is deficient in each lysosomal storage disorder?
|
Disease Name |
Enzyme affected |
Gene containing mutation |
Substrate that accumulates |
|
Tay Sachs |
Beta- hexosaminidase A |
HEXA |
GM2 Ganglioside |
|
Sandhoff |
Beta-Hexosaminidase A & Beta-Hexosaminidase B |
HEXB |
GM2 Ganglioside |
|
GM-1 |
Beta-galactosidase |
GLB1 |
GM1 Ganglioside |
|
Fabry |
Alpha-galactosidase A |
GLA |
Globotriaoslyceramide |
|
Gaucher |
Beta-glucocerebrosidase |
GBA |
Glucocerebroside |
|
Niemann-Pick Types A & B |
Acid-sphingomyelinase |
SMPD1 |
Spingomyelin |
|
Niemann-Pick Type C* |
|
NPC1 or NPC2 |
|
|
Pompe** |
Acid alpha-glucosidase (acid maltase) |
GAA |
Glycogen
|
*Mutations in the NPC1 or NPC2 genes cause a problem with the transport of lipids (fatty substances) in the cell. These lipids then build up in the cells, which causes cell dysfunction. There is further cell dysfunction because the lipids are not taken to the location where they are needed in the cell.
**Pompe is also a glycogen storage disorder.
Why are some lysosomal storage disorders more severe than others?
The severity of a Lysosomal storage disorder depends on the amount of enzyme activity that is present, and where in the body the substrate (debris) is accumulating. If some enzyme activity is present, symptoms usually begin later on (as in juvenile or adult onset version of the diseases.) The most severe disorders are where the debris builds up in the central nervous system, such as in Tay Sachs, Sandhoff and GM-1.
What techniques can be used to treat a lysosomal storage disorder?
To answer this question, please visit Technology Approaches.