All about Leukodystrophies Diseases


- Details
- Last Updated: Wednesday, 19 October 2016 09:32
Overview
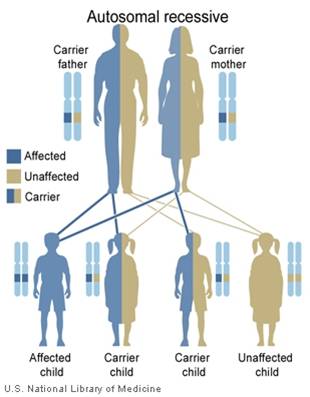
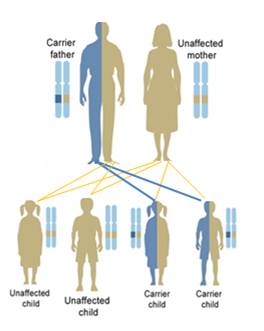
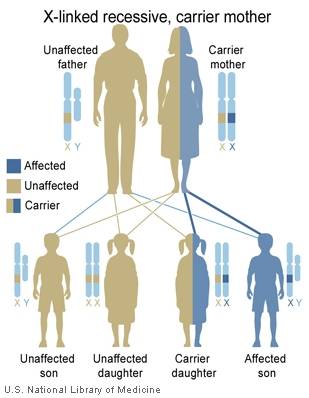
The leukodystrophies are a group of over 50 rare genetic disorders that affect the central nervous system by disrupting the growth or maintenance of the myelin sheath that insulates nerve cells. Myelin is commonly referred to as the brain's white matter. Myelin covers nerve cells and ensures the clear transmission of nerve impulses from one part of the body to another. These disorders are progressive, meaning that they tend to get worse throughout the life of the patient.
The word leukodystrophy comes from the Greek words leuko (meaning white), trophy (meaning growth), and dys (meaning ill). If you put these words together, the word leukodystrophy describes a set of diseases that affect the growth or maintenance of the white matter (myelin).
Leukodystrophies differ from multiple sclerosis (MS) in that leukodystrophies are caused by a defect in the genes involved with growth or maintenance of the myelin while MS is believed to be caused by an attack on the myelin by the body's own immune system.
Frequently Asked Questions
Which NTSAD diseases are leukodystrophies?
Canavan and Pelizaeus-Merzbacher are examples of leukodystrophies
What are leukodystrophies?
The leukodystrophies are a group of over 50 rare genetic disorders that affect the central nervous system by disrupting the growth or maintenance of the myelin sheath that insulates nerve cells. Myelin is commonly referred to as the brain's white matter. Myelin covers nerve cells and ensures the clear transmission of nerve impulses from one part of the body to another. These disorders are progressive, meaning that they tend to get worse throughout the life of the patient.
The word leukodystrophy comes from the Greek words leuko (meaning white), trophy (meaning growth), and dys (meaning ill). If you put these words together, the word leukodystrophy describes a set of diseases that affect the growth or maintenance of the white matter (myelin).
Leukodystrophies differ from multiple sclerosis (MS) in that leukodystrophies are caused by a defect in the genes involved with growth or maintenance of the myelin while MS is believed to be caused by an attack on the myelin by the body's own immune system.
How does a gene defect cause Canavan Disease?
All of our genes code for specific proteins, which have specific roles in our bodies. In Canavan disease, a defect in the ASPA gene causes an absence of a vital enzyme called aspartoacylase (ASPA.) An enzyme is a type of protein. The job of the ASPA enzyme is to break down N-acetylaspartate acid (NAA). When NAA builds up in the brain it leads to the symptoms of Canavan Disease.
It is sometimes helpful to think of the substrate and the enzyme as a lock and a key. The substrate is the lock, and the enzyme is the key. If the key is made incorrectly, it is not going to fit into the lock. An enzyme is a type of protein. In order for any protein to function correctly, it must contain the correct parts (which are called amino acids) and also be folded correctly.
The enzyme is made incorrectly if the gene that codes for the enzyme contains a spelling error, which is called a mutation. Mutations in the gene can cause the enzyme to contain incorrect parts, or cause it to be folded incorrectly. Sometimes the mutations in the gene cause the enzyme to not be made at all. In infantile Canavan disease, no enzyme is made. In juvenile/milder forms of Canavan disease, some enzyme is made, so there is less build up of NAA in the brain.
How does a gene defect cause Pelizaeus-Merzbacher?
All of our genes code for specific proteins, which have specific roles in our bodies. The PLP1 gene provides the code to build proteolipid protein 1, and a similar protein called DM20. These are the main proteins which are found in myelin. If the PLP1 gene contains a mutation, these proteins will not be made. If these proteins are not made then the myelin sheath cannot be formed correctly. This leads to the symptoms of Pelizaeus-Merzbacher disease.
What techniques can be used to treat Canavan Disease?
The answer to this question can be found on Technology Approaches.
Also, check out the latest Canavan Disease research.