
2017 Research Initiative Grants


- Details
- Last Updated: Monday, 29 May 2017 17:08
We are proud to announce the 2017 Research Initiative grant awards. Through an intensive review process, six were invited to submit full applications. After evaluations by Scientific Advisory Committee (SAC) members, a grant recommendation from a subset of the SAC group was made to NTSAD's Executive Committee for approval. As a result, three (3) grants have been awarded to the following researchers.
Alessandra Biffi, MD
Director, Gene Therapy Program
Dana-Farber/Boston Children’s Hospital Cancer and Blood Disorders Center
Boston, MA
Proof of concept study of
HSC gene therapy for Tay-Sachs disease
Goals of the Proposal
- To assess the feasibility and efficacy of an innovative gene therapy based on hematopoietic stem cells (HSCs) and lentiviral vectors (LVs) in the mouse model of Sandhoff disease (SD)
- To conduct a proof-of-concept study that could be translated into a larger scale study and future clinical development
Impact of the Research
The project will test a novel gene therapy approach for TSD and SD with the potential for clinical translation. This gene therapy works by establishing a stable population of brain cells, which will serve as a sustained and balanced supply of the deficient enzymes in the patients.
Dr. Biffi and her research team accumulated extensive experience in preclinical development and clinical translation of gene therapies for lysosomal storage disorders (LSDs) and TSD. The proposed study has a high likelihood of achieving transformative outcomes in TSD/SD due to the investigators’ expertise and the well-developed research plan.
Heather Gray-Edwards, PhD
Scott-Ritchey Research Center
Auburn University, Auburn, AL
Minimally invasive delivery of
AAV gene therapy in the Tay-Sachs Sheep
Goals of the Proposal
- To evaluate efficacy of adeno associated viral (AAV) gene therapy after cerebrospinal fluid (CSF) delivery in the sheep model of Tay-Sachs disease (TSD)
- To confirm previously proposed biomarkers of TSD, including MRI image, lipidomic signatures, cognitive testing, and neurologic evaluation
- To study the biodistribution and clearance of CSF delivered AAV gene therapy in the Tay-Sachs sheep
Impact of Research
Gene therapy, is one of the most promising treatment options in development.
The study will evaluate the efficacy and biodistribution of the state-of-the-art gene therapy in TSD sheep. Tay-Sachs sheep is one of the most relevant animal models of the disease, due to its large brain size and suitability for various biomarker studies.
The findings in this proposal will aid in expanding our knowledge on the treatment’s administration and biodistribution. The efficacy profile from the work with be necessary and helpful for future regulatory application before FDA. Most importantly, this study will generate useful data and insight about TSD biomarkers and contribute to future clinical studies in human patients.
Tim Wood, PhD, Greenwood Genetic Center. Greenwood, South Carolina and
Stephane Demotz, PhD, Dorphan, Lausanne, Switzerland
Development of a quantitative method for the determination of a pentasaccharide
in GM1-gangliosidosis patient cells to assess the potential therapeutic efficacy of a
beta-galactosidase pharmacological chaperone drug candidate
Goals of the Proposal
- To develop a quantitative, sensitive, and robust mass spectrometry based analytical method for the determination of a potential biomarker for GM1-gangliosidosis
- To explore treatment conditions of a drug candidate for GM1 in GM1-gangliosidosis patient cells using the analytical method developed in the first part of the project
Impact of the Research
GM1-gangliosidosis and mucopolysaccharidosis IVB (MPS IVB) are two diseases caused by beta-galactosidase deficiency. A biomarker that indicates the severity and progression of the disease and a convenient analytic method to determine the biomarker will significantly enhance the clinical trial design in the therapeutic development.
A preliminary study found a potential biomarker for both GM1 and MPS IVB. The level of the biomarker, a sugar molecule, was elevated by 10 to 100 fold in the urine samples of GM1 and MPS IVB patients than samples of normal donors. This compound has the potential to be utilized as a biomarker for future clinical trials.
The findings from the proposed work will develop a quantitative method to analyze the sugar molecule, validate its role as biomarker for the diseases. In addition, researchers will explore the optimal treatment conditions of an investigational pharmacological chaperone drug candidate for GM1-gangliosidosis.
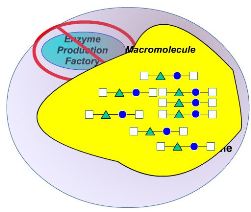 Overview
Overview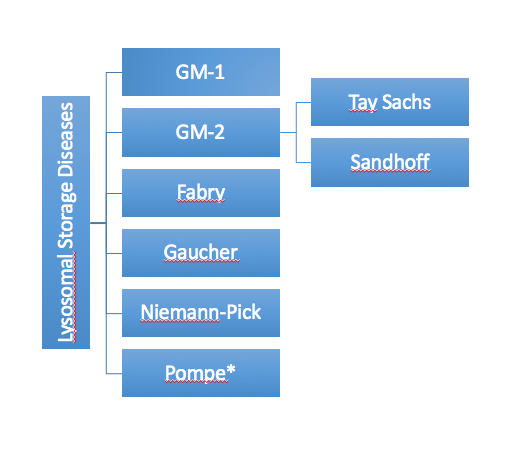
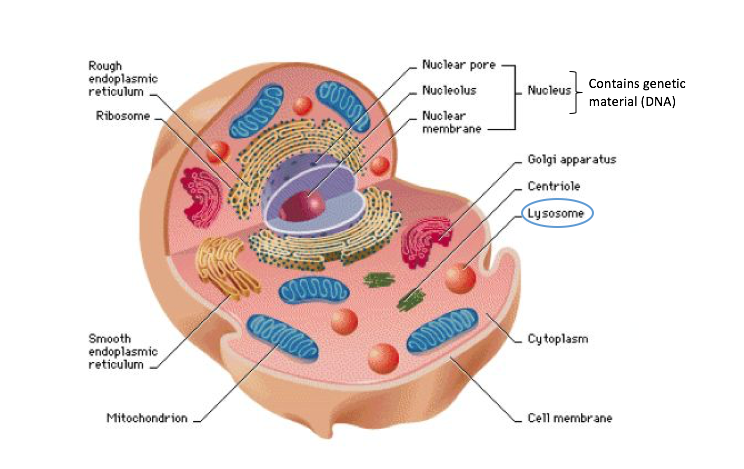
 Join us for a fun filled day of a 1-mile walk, silent auctions, raffles, lunch and door prizes. All proceeds will be donated to
Join us for a fun filled day of a 1-mile walk, silent auctions, raffles, lunch and door prizes. All proceeds will be donated to 